|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
기술문서. ISO 13485. Risk management 등 의료기기 유럽 CE MDD 인증 컨설팅 전문가가 쳬계적이고 빠르게 인증지원해 드립니다. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Medical Device Directive (MDD; 93/42/EEC) 에는 체외 진단 지침 ( In Vitro Diagnostics, IVD) 또는 Active Implantable 의료 기기 (AIMD)에서 포함되지 않는 모든 의료기기가 그 범위에 속합니다. MDD는 의료 기기의 평가를위한 필수요건 (essential requirements), 분류규칙(classification rules) 및 적합성 경로 (conformity routes for assessment) 에 대해서 복잡하게 설명하고 있으며 의료기기에 관한 구체적인 결정은 MDD의 23 항목, 많은 첨부문서 (annexes) 그리고 18 가지의 분류규칙의 이해를 필요로 합니다.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
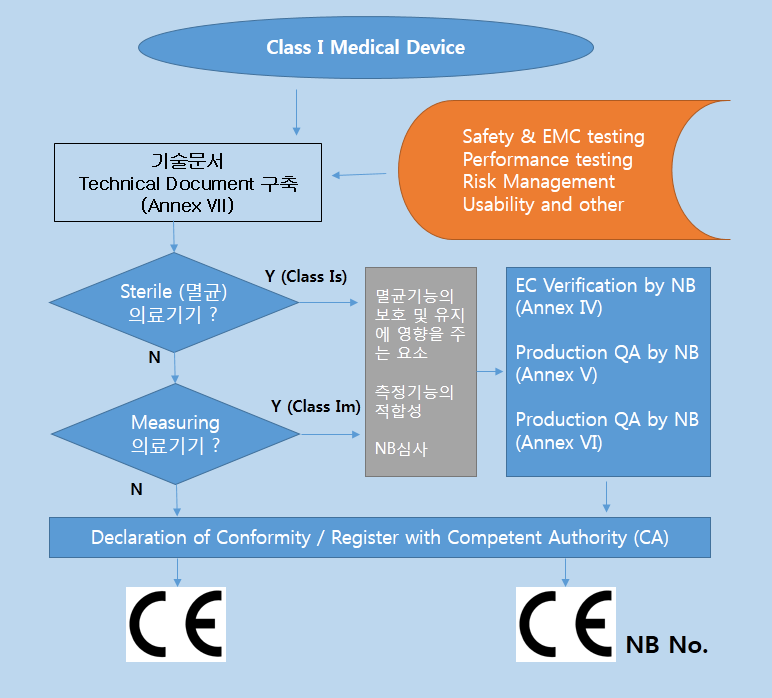
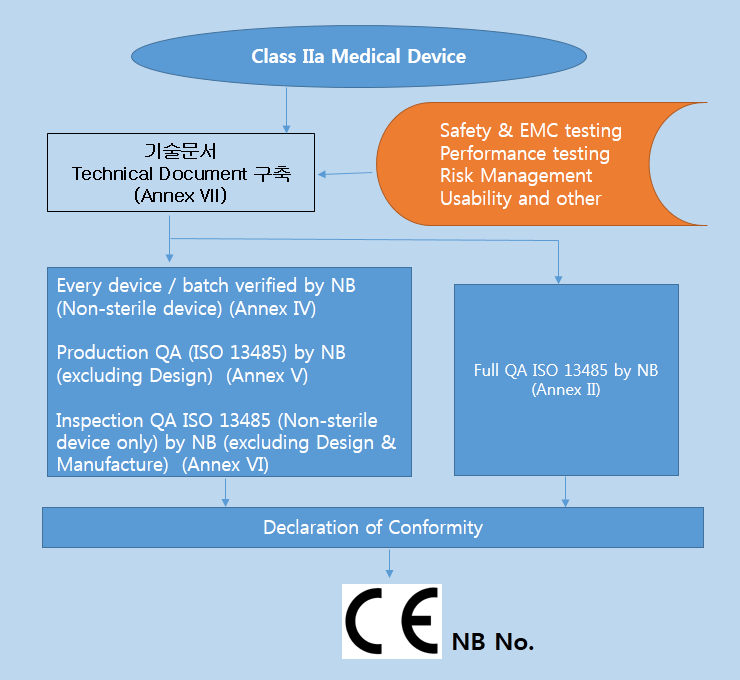
MDD 에서는 의료기기의 등급을 위험등급에 따라 Class I, Class Im (측정기능), Class Is (멸균대상기기), Class IIa, Class IIb, Class III 로 분류한다. 등급은 기초적인 검토과정을 거치고 MDD 의 Annex IX 의 Rule 1~18 에서 해당되는 의료기기를 찾아서 등급을 결정하면 된다..
해당 의료기기의 사용시간을 정하는데 사용기간을 아래 3가지 중에서 선택한 후 대상 의료기기가 신체내부에 삽입이 되지 않고 전원공급이 필요없는 의료기기라면는 Rule 1~4 사이에 따르면 된다.
1. Transient – 일시적 (보통 사용기간이 지속적으로 60분 미만) 2. Short term - 단기간 (보통 사용기간이 지속적으로 30일 미만) 3. Long term – 장기간 (보통 사용기간이 지속적으로 30일 이상)
1. Non-invasive devices (비삽입기기) Rule 1 ~ Rule 4
2. Invasive devices (삽입기기) Rule 5 ~Rule 8 의료기기가 삽입되는 경우 원래 가지고 있는 곳 (눈, 콧구멍, 입, 귓구멍 등) 을 통해서 삽입하는 것인지 아니면 수술등을 통해서 삽입하는 것인지 구별 해야 하며. 원래 갖고 있는 곳을 통해서 삽입한다면 Rule 5번에 따른다. 수술을 통해서 삽입하는 기기는 그 사용기간이 일시적이냐 단기간이냐 장기간 인가에 따라서 Rule 6, Rule 7, Rule 8으로 나눈다.
3. Additional rules applicable to active devices (능동기기에 적용하는 추가 Rules) Rule 9 ~Rule 12 의료기기가 밧데리 또는 전원 플러그로부터 에너지 공급을 받는다면 Rule 9~12에 따른다
4. Special Rules (특별분류) Rule 13 ~Rule 18 그외 특별히 사용되는 의료기기는 Rule 13~18번에 따르지만, 기기가 Rule 1~12에 속한다고 하여도Rule 13~18에 속하게 되면 Rule 13~18을 등급을 따른다.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
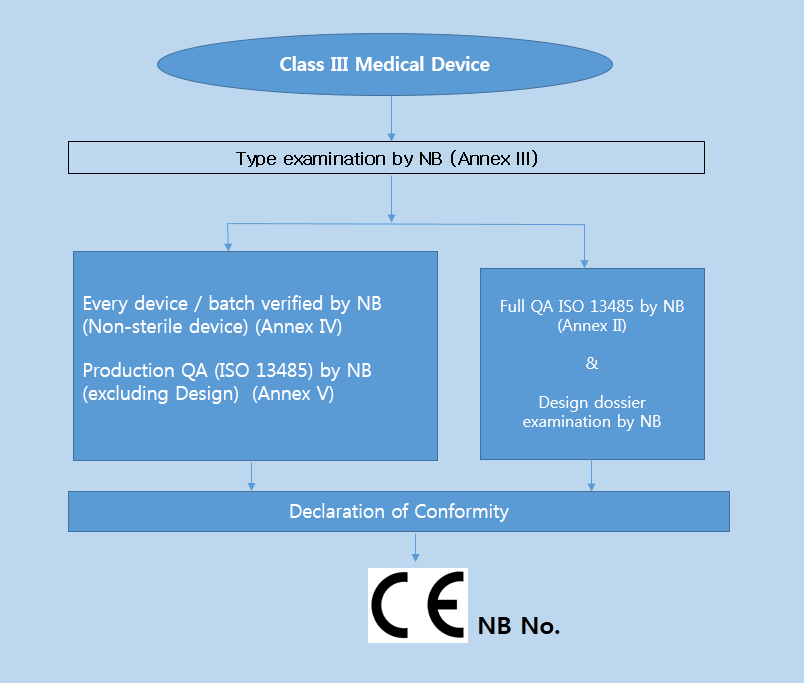
의료기기의 위험정도에 따라서 등급분류를 하여 그에 적합한 적합한평가절차가 다음과 같이 결정된다.
Class IIa 및 Class IIb에 관련되는 기술문서 (Technical Documentation) 는 Annex II, V 의 인증기관 (Notified Body) 에 의해 검토되어야 한다.
* 부속서 II : EC 적합성선언 (종합적인 품질보증시스템 - Full Quality Assurrance System) 제조자는 해당제품에 대하여 3항에 정한 설계, 제조, 최종검사에 관해 승인을 받은 품질시스템(ISO 13485)을 적용하여야 한다. 또 3.3항 및 4항에 정한 심사를 받고, 5항에 정한 EC사후관리를 받아야 한다
* 부속서 III : EC 형식시험 (EC TYPE-EXAMINATION) EC 형식시험은 지침의 관련규정에 만족하는가에 대하여 대표적으로 생산제품의 샘플로써 검사, 확인하는 순서이다
* 부속서 IV : EC검증 (EC VERIFICATION) EC검증은 4항의 순서에 따른 제품이 EC의 형식시험 인증서에 기재된 형식에 적합하고, 또 이 지침의 해당요건에 적합한 사실을 제조자 또는 EC지역 내 대리인이 보증하고 선언하기 위한 절차이다
* 부속서 V : EC 적합성 선언서 (제조품질보증 - Production Quality Assurance) 제조자는 관련제품의 제조에 관하여 품질시스템을 적용하고 3항에 규정한 최종검사를 실시하며, 4항에 서술한 EC 사후관리 감시를 받아야 한다.,
* 부속서 VI : 적합성 선언서 (제품품질보증- Product Quality Assurance) 제조자는 3항에 지정하는 제품검사 및 시험에 관해 승인을 필한 제품의 품질시스템을 운용하고 4항에 서술한 심사를 받는다.
* 부속서 VII : 적합성 선언서 EC적합성 선언서는 2항에서 부과하는 의무를 다하는 제조자 또는 그 EC 내 대리인이 해당제품이 이 지침의 적용규정을 만족하고 있음을 보증하고 선언하기 위한 절차이다. 해당제품이 멸균 및 측정기능이 있는 경우 5항에서 부과하는 의무를 다하는 제조자 또는 그 EC 내 대리인이 마찬가지로 하여야 한다.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
의료기기는 의료기기지침의 부속서 1 에서 요구하는 필수요구사항을 만족시켰는지 검토 되야 한다.
1. 일반요구사항 기기는 어떤 조건 하에서 특정 목적을 가지고 사용될 경우 진료 조건이나 환자 또는 사용자의 안전과 건강 등을 손상하지 않도록 설계되고 제조되어야한다. 또한 그 기기를 사용할 때 예상되는 위험이 환자의 이익에 불리하게 작용할 경우 그 위험이 수용 가능한 위험이어야하고 환자의 건강과 안전에 위태롭지 않도록해야한다. 제조 업체가 고안한 기기의 설계와 구조는 안전 원칙들을 준수해야만하며 일반적으로 알려진 기기 종류의 상태를 고려해야한다 가장 적합한 해결책을 선별하기 위해 제조 업체는 다음 원칙을 준수해야한다.
* 제조 업체가 고안한 기기의 설계와 구조는 안전 원칙들을 준수해야만하며 일반적으로 알려진 기기 종류의 상태를 고려해야한다 * 가장 적합한 해결책을 선별하기 위해 제조 업체는 다음 원칙을 준수해야한다. * 가능한 한 위험을 제거하거나 피해야한다. (안전한 설계 및 구조) 피할 수없는 위험의 경우 필요하다면 "경고"기능을 포함한 적합한 예방 조치를 취해야한다. * 사용자에게 예방 조치의 부족으로 야기되는 그 외의 위험에 대해서 알려야한다. * 기기는 제조 업체가 의도한 성능을 수행해야하고 제조 업체가 명시한 바와 같이 1 조 (2) ()에 명시된 하나 이상의 기능에 적합한 방식으로 설계, 제조, 포장되어야한다 * 항목 1, 2 및 3에 명시되어있는 기기의 특성과 성능은 기기 작동 중에 장애가 발생할 경우 제조 업체가 명시한 기기의 수명 한도 내에서 진료 조건, 환자 및 타인의 안전에 해를 끼쳐서는 안된다. * 기기는 제조 업체가 제공한 정보 및 지시 사항에있어서 기기의 운반 및 보관시 그 특성과 성능이 손상되지 않도록 설계, 제조, 포장되어야한다. * 의도된 작업에 반하는 예기치 못한 부작용은 수용 가능한 위험이어야한다
2. 설계 및 구조 관련 요구 사항
- 화학적, 물리적 및 생물 학적 특성 - 전염 및 세균 감염 - 구조 및 환경적 특성 - 측정 기능이 장착된 기기 - 방사능 으로부터의 보호 - 에너지원에 연결되거나 장착된 의료 기기 요구 사항 - 제조 업체 및 진찰 기록에 의해 제공되는 정보
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
임상 시험용 의료 용구의 경우 제조자 또는 그 EC는 지역 내의 대리인은 부속서 V Ⅲ에 말하는 절차에 따라 임상 시험을하는 가맹국의 행정 당국에 신고하여야한다. CLASS Ⅲ의 의료 용구 및 매입 식이고 장기 침습 의료용 구로 CLASS Ⅱ 또는 Ⅱ B의 의료 용구의 경우 제조자는 신고한 후 60 일 뒤에 임상 시험을 개시할 수있다. 단, 행정 당국이이 기간 내에 공중의 건강 또는 공공 정책에 대하여 고려한 결과, 시험을 허가하지 않는 뜻을 제조자에 통고한 경우를 제외한다.
그러나 윤리위원회가 그 시험 프로그램에 찬성 의견을 표명한 경우 가맹국은 제조자에 대하여 상기 60 일간의 기한 만료 이전이라도 제조자에 대하여 임상 시험 개시를 허가할 수있다. 제 2 항에서 말하는 의료 용구 이외의 의료용 구인 경우 윤리위원회가 그 시험 프로그램에 찬성 의견을 표명했을 때는 가맹국은 제조자에 대하여 통고일 직후부터 임상 시험의 개시를 허가 할 수있다.
임상 시험은 부속서 X의 조항에 따라 실시되어야한다.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||






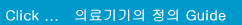






![]()
