![]()
|
메가써티스아이엔씨 전 세계 인허가 컨설팅 전문 고객지원 Tel: 02-2647-7632, e-mail : help@certis.co.kr
|
|
|
||||
| |
|
|
|
|
|
|
|
|||
|
|
|
|||
|
|
||||
|
제조허가. 수입허가, GMP구축. 기술문서. 위험관리. SW밸리데이션. 사용적합성. 멸균밸리데이션. 전기기계적안전성. 전자파. UDI. GLP ... |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
의료기기법에 따라 의료기기를 제조하는자는 의료기기 품질경영시스템을 구축하고 수행하는데 필요한 품질문서 및 기록을 관리해야 하며 GMP는 의료기기의 설계. 개발. 제조. 시판후 관리 등 전과정에 대한 품질시스템의 확보를 통해 안전하고, 유효하며, 의도된 사용목적에 적합한 품질의 제품을 일관성 있게 제조한다는 것을 국제규격과 조화된 방법으로 평가되어 인증을 받는 것입니다.
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
의료기기 GMP 품질관리 적용대상
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
의료기기 GMP 심사의 구분
의료기기 GMP 심사기준
의료기기 GMP 심사기준의 병행적용 일정
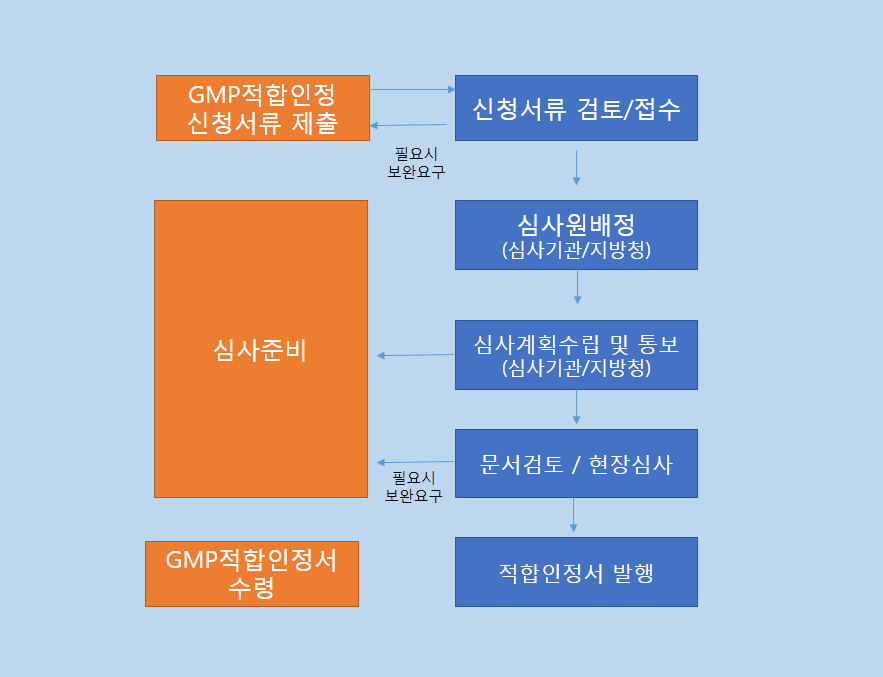
의료기기 GMP 심사절차
의료기기 GMP 제출서류
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
제조허가. 수입허가, GMP구축. 기술문서. 위험관리. SW밸리데이션. 사용적합성. 멸균밸리데이션. 전기기계적안전성. 전자파. UDI. GLP ...
|

 .
.